User guide
Steps to follow
1. Input query motifs.2. Input source sequences.
3. Input other mandatory inputs.
Reference position
Choose Strand
Threshold frequency
Block size
4. Case study
Using MotDet, the following three basic steps can be used to analyse the given query sequences for motifs. Those steps are as follows:
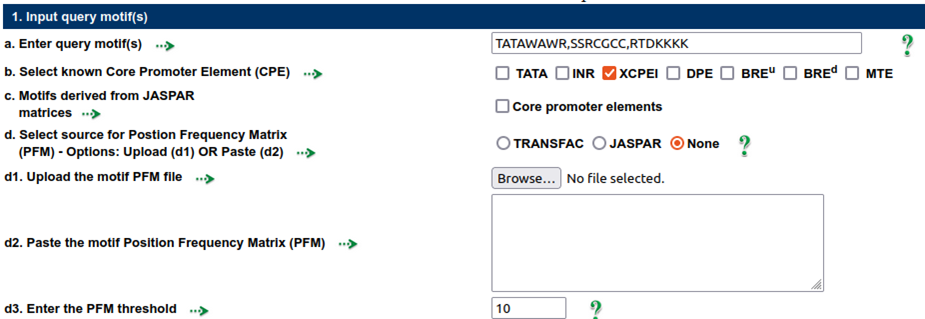
Custom DNA motifs with lengths ranging from 5 to 20 can be entered by the user.
These motifs may take the form of consensus sequences or specific patterns.
The IUPAC format should be followed for consensus motifs.
The list of symbols used in IUPAC format is as follows:
IUPAC Format
|
Symbol
|
Represents what?
|
Why this symbol?
|
|
G
|
G
|
Guanine
|
|
A
|
A
|
Adenine
|
|
T
|
T
|
Thymine
|
|
C
|
C
|
Cytosine
|
|
R
|
G or R
|
puRine
|
|
Y
|
T or C
|
pYrimidine
|
|
M
|
A or C
|
aMino
|
|
K
|
G or T
|
Keto
|
|
S
|
G or C
|
Strong interaction (3H bonds)
|
|
W
|
A or T
|
Weak interaction (2H bonds)
|
|
H
|
A or C or T
|
not-G, H follows G in the alphabet
|
|
B
|
G or T or C
|
not-A, B follows A
|
|
V
|
G or C or A
|
not-T(not U as it stands for uracile), V
follows U
|
|
D
|
G or A or T
|
not-C, D follows C
|
|
N
|
A or G or C or T
|
aNy
|
Input example for custom DNA motif: TATAWAWR,RGWVY
The user can choose from a list of predefined motifs.
Following a thorough literature search for a few mammalian species, this list was created.
A motif position frequency matrix (PFM) that has been imported from either TRANSFAC or JASPAR is available from the user.
Input example for Jaspar motif PFM matrix:
>MA0003.1 TFAP2A
A [ 0 0 0 22 19 55 53 19 9 ]
C [ 0 185 185 71 57 44 30 16 78 ]
G [185 0 0 46 61 67 91 137 79 ]
T [ 0 0 0 46 48 19 11 13 19 ]
Input example for TRANSFAC motif matrix:
>M2-V$TBP_Q6
1 1 1 17
0 0 0 20
0 0 0 20
15 2 1 2
2 0 0 18
12 3 2 3
2 8 5 5
The given matrices will be transformed into consensus sequences by MotDet before being used to analyse query sequences. Multiple motifs PFM in a file can be uploaded or pasted. The PFM threshold can be used to determine how strictly the matrix is converted to the consensus sequence. The IUPAC symbol is assigned for a position based on the PFM threshold, which restricts the information content for that position.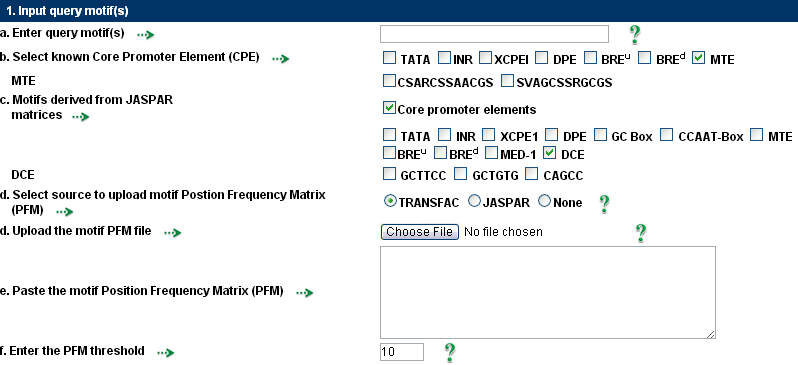
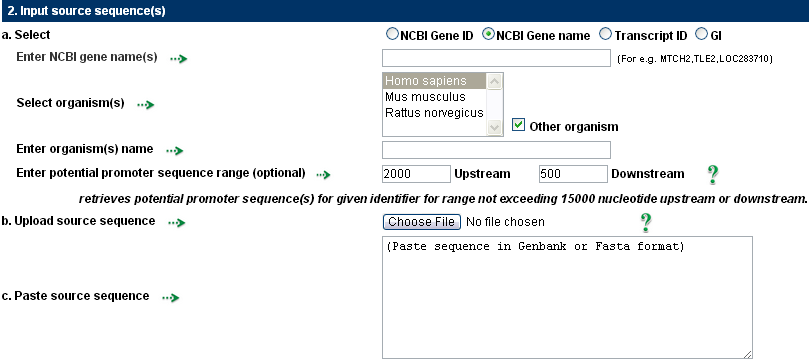
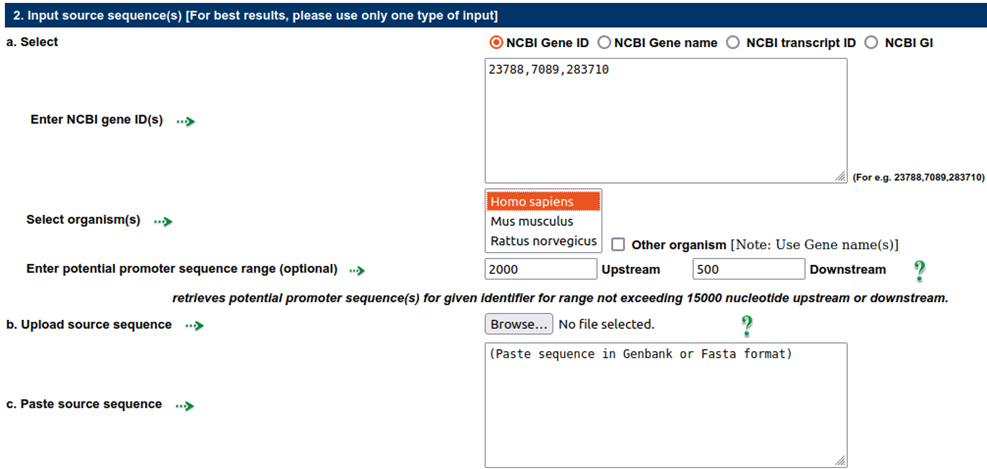
Nucleotide sequences that will be examined for user-defined motifs should make up the query sequences. This input can be provided in six different ways: gene ID, gene name, transcript ID, GI, paste and upload the sequence. To analyse their potential promoter sequences, enter an NCBI gene ID, gene symbol, transcript ID, or GI list separated by a comma. From NCBI, sequence data is downloaded and kept locally in a database. All possible promoters are taken into account when a gene ID or gene symbol is input in order to retrieve potential promoter sequences.
When a gene name is entered as an input, the user has the option of specifying one or more different organisms. The user can use this to perform motif analysis on genes that are orthologous. By providing transcript ID or GI, the user can analyse each potential promoter sequence of the desired genes individually.
Input example for Gene ID: 23788,7089,283710
Input example for Gene name: MTCH2,TLE2,LOC283710
Input example for Transcript ID: NM_014342,NM_003260,NM_001243538
Input example for GI: 223941821,221219046,343780888
The range of the potential promoter sequence can be changed according to the needs, with a limit of -15000 to +15000.
The sequence in FASTA or Genbank format can also be uploaded or pasted by the user.
Click Genbank or FASTA, for instance FASTA or Genbank
With query sequence input up to 1 MB, software functions well.
3. Input other mandatory inputs
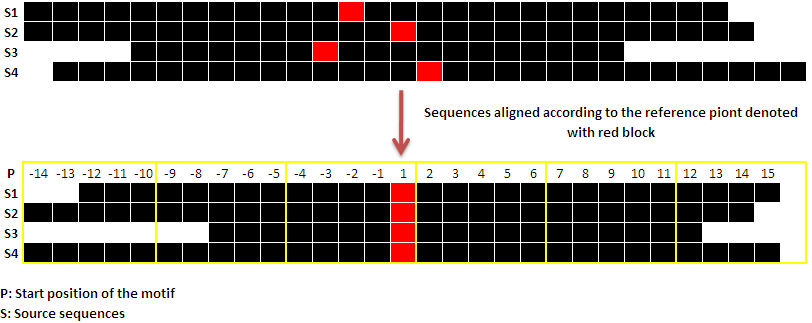
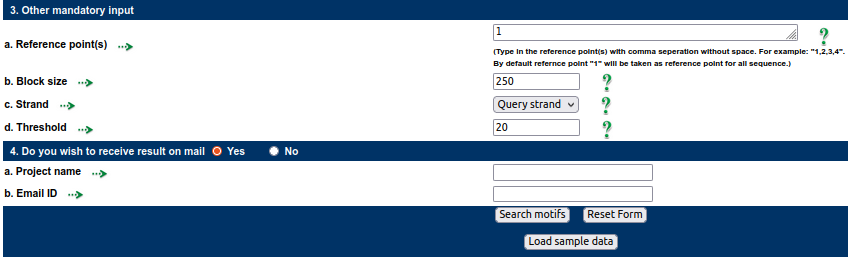
Reference position: Blocks of nucleotides are used to divide up the submitted query sequences. The sequences are aligned to one another with respect to a position, such as the gene's reference point, before being divided. This type of sequence alignment would help indicate uniform sequencing upstream and downstream, which would facilitate block-wise analysis of the sequences for motifs. In light of this, reference points are crucial in the blockwise analysis of motifs. The transcription start site obtained from dbTSS will be used as the reference point when the user provides a gene ID or gene name. Additionally, the "start position" of the sequence is used as the default reference point when users upload or paste sequences; however, this reference point can be changed based on the needs of the user.
The source sequences are aligned with respect to their reference point in the following pictorial representation. The sequences are shown as black lines. The red block designates the reference point, and yellow lines define the blocks of the sequences.
Choose Strand: One or both strands may be chosen by the user for motif analysis. Separate results will be provided for the complementary strand and the query.
Threshold frequency: The minimum number of source sequences (expressed as a percentage) in which a specific motif is anticipated to be present is referred to as the threshold. The user may change the threshold value from its default of 25 if necessary.
Block Size: The sequence will be broken up into smaller blocks of sequences in order to find the common variants and co-variants. The block size will be used to determine the size of such blocks (in units of base pairs). Therefore, common variants and co-variants will be found with a lower mean deviation when the block size is smaller, and vice versa. 250 nucleotides is the block size's default value and it can be altered by user's choice.

The user can perform a blockwise analysis of the sequence for the motifs by providing the necessary inputs. A web browser will display all of the results. The result can be downloaded using the link at the bottom of the result page, or the user can specify a "Project Name" and "Email ID" to receive the result on a specific email ID.

Query motifs:The following query motifs were used to analyze the over-representation of the motifs and the variants: TATAWAWR, SSRCGCC, and RTDKKKK from query motifs, and XCPEI from core promoter elements (CPE)
Source Sequences: As source sequences, the promoter sequences of three NCBI IDs—23788, 7089, and 283710—from 2000 to 500 nts upstream to downstream w.r.t. TSS are employed.
Other Mandatory inputsReference point -1 (in this case, 1 denotes the TSS of human promoter sequences), block size 250, strand query, and threshold 20 were the default values used.
Outcomes of case study
When your mouse pointer hovers over the input button on the result page, the provided inputs are shown.

Five IDs are displayed here. Three transcripts with the NCBI gene ID 7089 exist: 3047633, 3029612, and 3029165; the promoter sequences for these transcripts are retrieved and taken into account for analysis.
Significant motifs and the variants
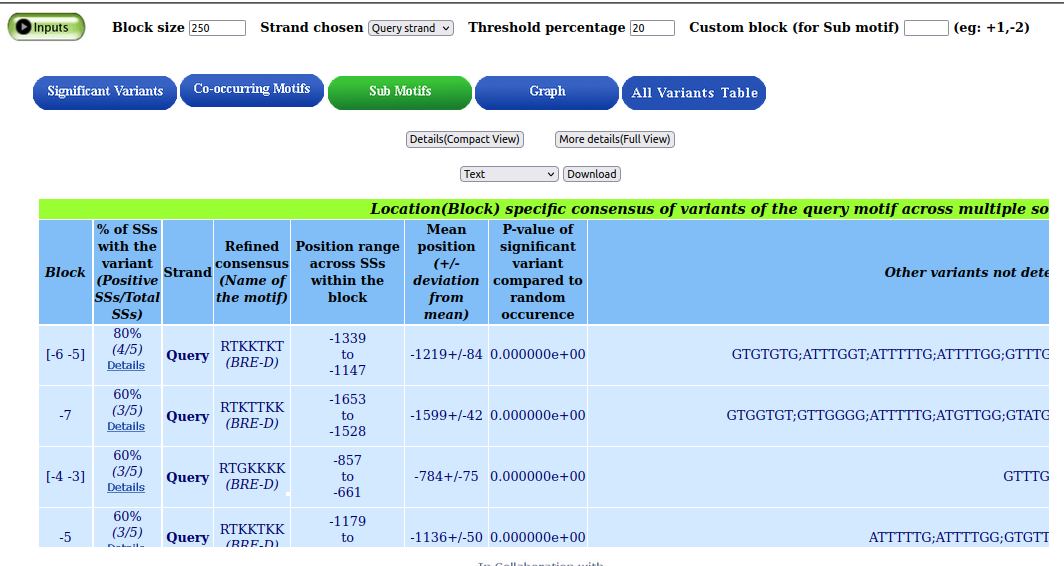
BRE-D, RTDKKKK and ATTTTTT are the overrepresented motifs and variants.It displays the position range in the block, the p-value, and the position of variants in the provided source sequences.

A pictorial representation for detection of common variants
Co-occuring motifs and the variants
The variants are displayed, and the overrepresented co-occurring motifs are BRE-U (SSRCGCC) and BRE-D (RTDKKKK). Additionally, the "significant covariants" button for each query motif provides access to the co-occurring variants of each query motif.
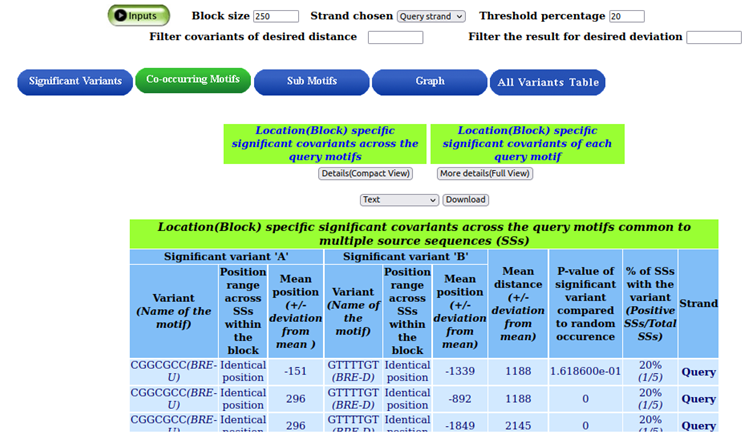
A pictorial representation for detection of cooccuring variants
Submotifs
The derived subconsensus of significant motif variants (BRE-D (RTDKKKK)) is represented by the blocks RTKKTKT, RTKTTKK, RTGKKKK, and RTKKTKK.
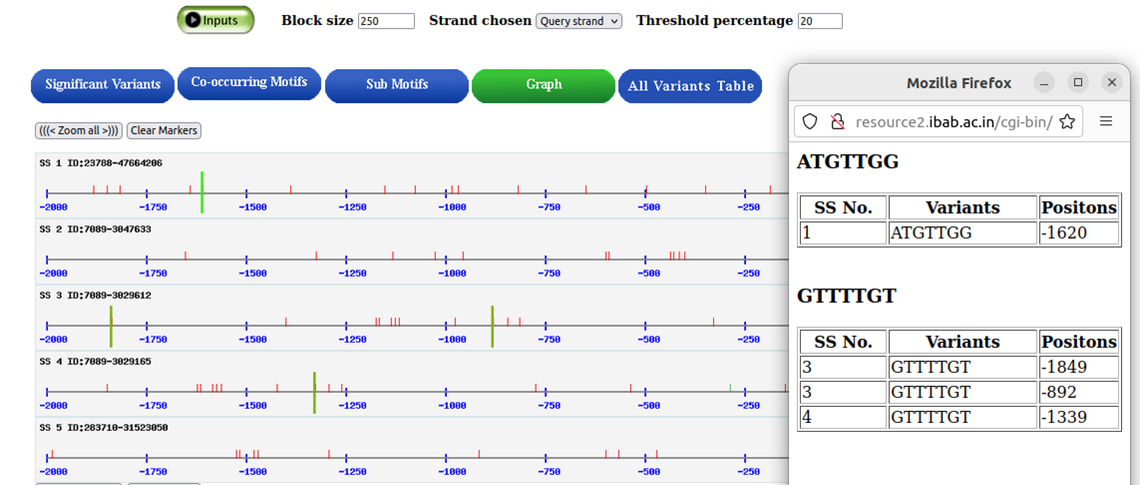
Graphical summary of the variants
A line graph displays every variant found in each of the source sequences.By clicking on the red vertical line markers, the variants and the positions are shown with highlighted markers.
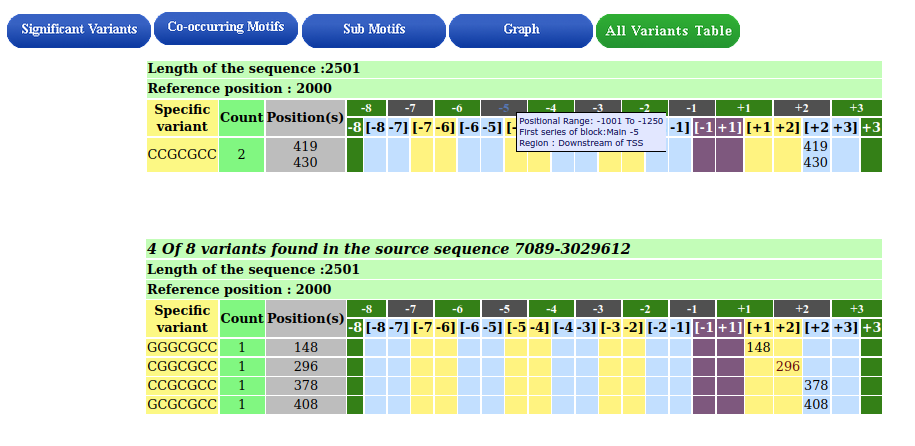
Tabular summary of the variants
The complete set of variants for each source sequence is displayed. The table has two rows: the first row displays non-overlapping blocks with mouse-over details, and the second row displays overlapping blocks. Each of the queried motifs' variants and locations are displayed.